Introduction
|
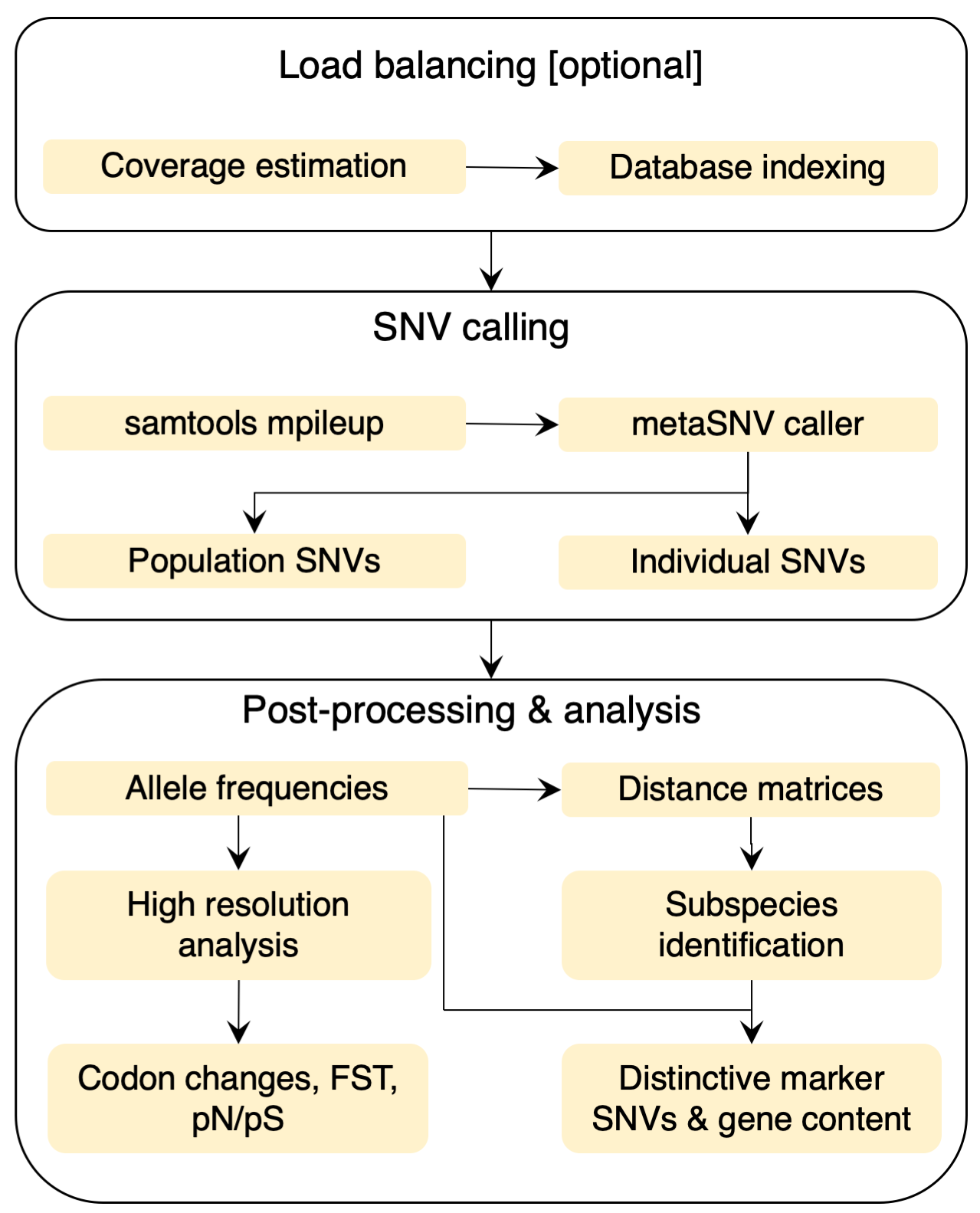
MetaSNV is a pipeline for calling metagenomic single nucleotide variants (SNVs). It was designed to scale well with the exponentially increasing amount of available metagenomic datasets and is capable of handling large multi-species references. Input: MetaSNV takes a list of alignment files (BAM) as input. Each should represent one metagenomic sample aligned to a reference genome collection. The reference can be any multi-sequence FASTA file encompassing thousands of bacterial species. There should be one reference per species. Output: MetaSNV differentiates two classes of variants: population SNVs (pSNVs) and individual SNVs (iSNVs). The former are a non-reference nucleotide observed in more than 1% of all reads combined across all samples. If multiple different non-reference nucleotides are above this frequency, all are reported independently. The individual variants are those that fall below the 1% frequency population threshold, but are confidently observed in at least one sample. As such, positions that have a within-sample frequency above 10%, in a sample where that position is covered by at least 10 reads, are considered to be individual variants. Analysis: MetaSNV offers scripts for the analysis of the output. Taxon, sample and position filters are applied post SNV calling. For downstream processing, pair-wise distance matrices are computed between all samples. Based on these distances, subspecies can be identified using robust cluster detection. SNVs and genes distinctive of these subspecies can then also be detected. |
|---|
Further Information
Download and InstallationPlease see the metaSNV GitHub page for downloads and installation instructions.Getting startedTo get started with metaSNV v2, please refer to the updated manual and the GitHub page.Citing metaSNV
|